Predictive computational methods
Contents
3.4. Predictive computational methods¶
The low cost of sequencing means that databases of sequences have been expanding very rapidly in comparison to other information, which is much harder to determine. Computational predictive methods aim to predict structure or function from sequence in order to bridge this gap. Here I describe some of the challenges and methods in this space, many of which leverage the ontologies and databases described in the previous sections.
3.4.1. Prediction tasks: Protein classification prediction¶
As previously mentioned, proteins are often classified by structural similarities. This information is often used because researchers identify a gene of interest, but information about it’s function or structure (in PDB) has not yet been captured and stored (i.e. the protein is “uncharacterised”). In such cases, it’s often necessary to make inferences about protein structure or function based on their similarity to known proteins. This is sometimes done using sequence similarity (e.g. BLAST), but sequence similarity can vary considerably between proteins with the same underlying structure. This is why structural similarity searches based on protein classification are preferred.
3.4.1.1. SCOP¶
The Structural Classification of Proteins (SCOP) database[38] classifies all proteins with known structure based on their structural similarities, based on the consideration of the protein’s constituent domains. The classification is mostly done at the level of families, superfamilies, and folds arranged in a tree structure. Families represent the most similar proteins, which share a “clear evolutionary relationship”, while superfamilies represent less close evolutionary relationships, and folds represent the same secondary structure. This protein classification task, while aided by automation, was carried out largely by manual visual inspection.
SCOP was updated until 2009, but has been succeeded by SCOP2[115]. However, SCOP2 has a different underlying classification system, based on a complex graph, rather than a hierarchy. The CATH (Class, Architecture, Topology, Homologous superfamily)[116] database provides another classification system, which operates hierarchically, but is created mostly via automation, which leads to major differences between the classifications[117].
3.4.1.2. SUPERFAMILY¶
SUPERFAMILY[118] is structural annotation procedure and associated web resource, which uses Hidden Markov Models (HMMs) to assign sequences to SCOP domains, primarily at the superfamily level. This is a process analagous to gene annotation.
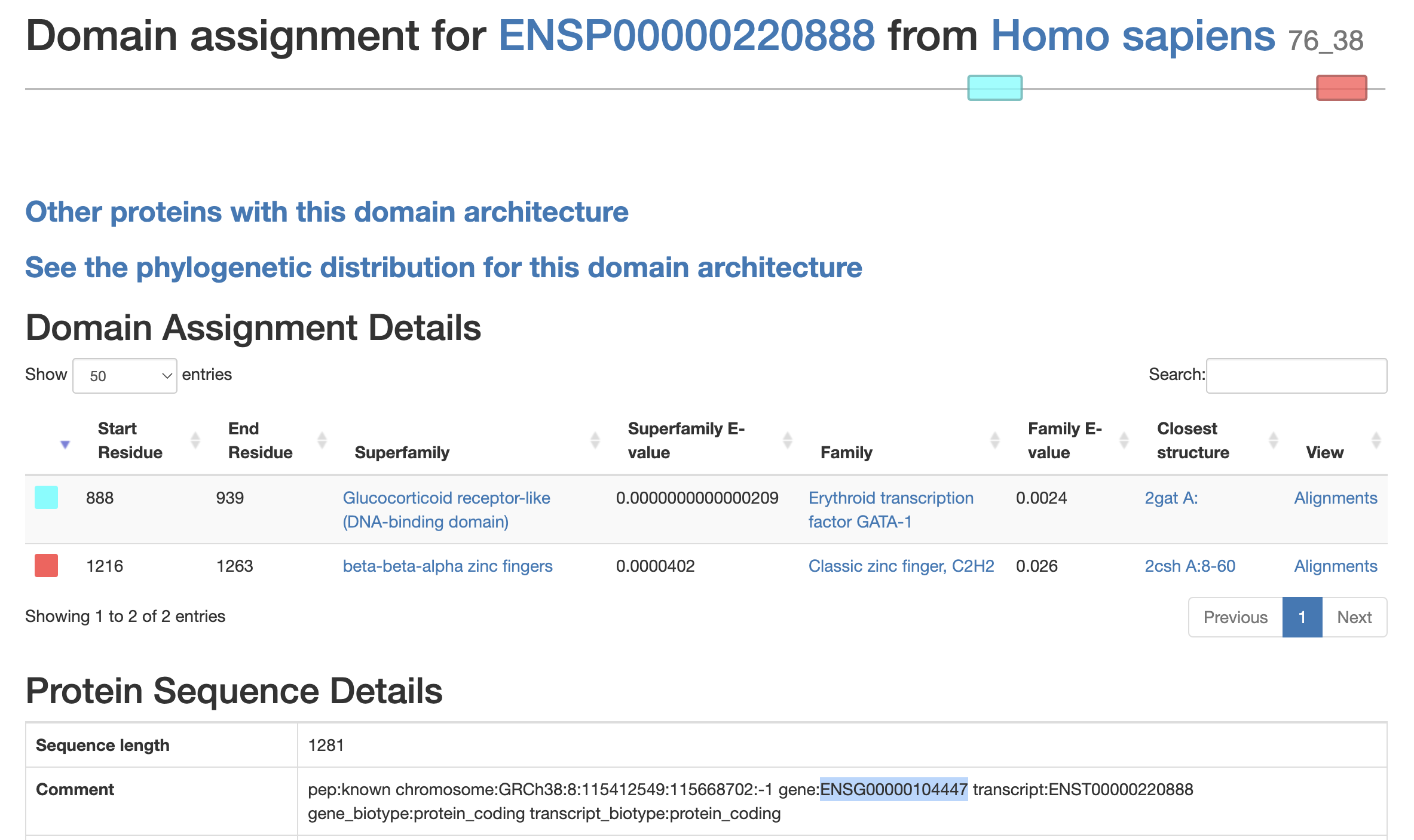
Fig. 3.5 Domain assignments for ENSP00000220888.5 - a human protein.
The image shows the protein is mostly not made up of protein domains, but has two domains assigned, highlighted in blue (Glucocorticoid receptor-like (DNA-binding domain)) and red (beta-beta-alpha zinc fingers).¶
This domain assignment is also used as a form of functional annotation and allows the functions of poorly understood proteins to be inferred based on how closely they match known superfamilies. This is how the website is frequently used - as it allows users to search a new sequence and see which domains are predicted to fall in the sequence and where. It is also possible to see domain assignments of known proteins, e.g. by ENSP (Ensembl Protein ID), see Fig. 3.5.
HMMs are very successful at such assignments since pairwise correlations between proteins (or their domains) and other proteins in the family may be weak, but consistently for many proteins; this can be picked up by an HMM. The superfamily level is chosen since it is the broadest level which suggests evolutionary relationships, but SUPERFAMILY also generates assignments at the (stricter) family level.
HMMs are created by first finding closely relating protein homologs for a given protein superfamily using BLAST, and then extending it by comparing the HMM to more distantly related homologs. The resulting HMM library is fine-tuned by some manual curation.
Although SUPERFAMILY’s primary resource is it’s HMM library, it also integrates a range of other tools for sequence analysis, for example protein disorder prediction (D2P2) and GO annotation (dcGO), as well as a domain-based phylogenetic tree (sTol). In addition, SUPERFAMILY makes available all sequences that it uses to build HMM models, some of which cannot be found elsewhere.
SUPERFAMILY update
Contributions in this section
The SUPERFAMILY website and resources were jointly maintained between members of Computational Biology group (then) at Bristol, which involved replying to user emails, and updates to the website.
In 2014, a SUPERFAMLIY update paper was published. I contributed by editing the paper and adding a small number of proteome sequences in the class Cyanophyceae - 5 of the proteomes available on SUPERFAMILY.
I contributed to SUPERFAMILY’s 2014 update[3]. The SUPERFAMILY database of proteomes doubled from 1400 to over 3200 from 2010 to 2014, containing sequences from across the tree of life, including 1714 species and 1544 strains. The update paper described this development, as well as highlighting SUPERFAMILY as a resource for unique proteomes that are not found elsewhere (e.g. Uniprot), and describing the update to the (at the time) most recent human reference genome.
3.4.2. Prediction tasks: Protein function prediction¶
Human genes can have multiple functions, but currently, we don’t even know one function for all of them. Although we have their sequence, some genes are completely functionally unknown to us. Protein function prediction is the task of predicting protein function (usually in terms of ontology terms) from protein sequence.
3.4.2.1. DcGO¶
The aim of the domain-centric Gene Ontology (dcGO)[119,120] tool is to give insight into uncharacterised or poorly characterised proteins by leveraging information about the content of their constituent protein domains. It annotates domains and combinations of domains (a.k.a. supradomains) to phenotype terms from a variety of ontologies, including the Gene Ontology (GO), Mammalian Phenotype ontology (MP), Disease Ontology (DOID), Zebrafish ontology (ZFA). Domain information comes from SUPERFAMILY, and annotations between (supra)domains and phenotype terms are made below a cut-off of FDR-adjusted statistical associations between the entities. Using phenotypes from a range of species serves to make use of greater numbers of experiments, and therefore increases the number of little-known proteins across species that DcGO can make predictions about.
3.4.3. CAFA¶
Critical Assessment of Functional Annotation[5,121,122] (CAFA) is an international community-wide competition for the prediction of protein function, which aims both to stimulate research in the field of protein function prediction, and to measure progress in the field. It has been running approximately every 2-3 years since 2013.
Each CAFA challenge begins by the organisers releasing a large number of target sequences (over one hundred thousand) across multiple species, about which participant teams must make predictions. After the competition closes, the organisers wait 3 months, by which time, new experimentally verified protein functions will be found (representing ~3% of sequences in past competitions) and these are the data set against which the predictors are measured.
Participants can use any additional data they see fit to make predictions, which must be triples containing a sequence ID, ontology term ID (e.g. a GO/HP identifier), and a confidence score between 0 and 1. A score of 1 indicates a completely confident prediction, while a score of 0 is equivalent to not returning the prediction. Each team may submit up to three models, the best of which is ranked.
The target sequences consist of a mixture of “no-knowledge” and “limited-knowledge” sequences. No-knowledge sequences are sequences which upon release have zero experimentally-validated GO annotations to any of GO’s three constituent ontologies (biological process, cellular component, and molecular function). Limited-knowledge sequences are sequences with one or more annotations in one or two GO ontologies, but not all three.
3.4.4. Prediction tasks: Variant prioritisation¶
Variant prioritisation is a version of protein function prediction in which long list of genes or variants (obtained for example through a GWAS experiment) are narrowed down to a shorter list of variants or genes which are more likely to be causal.
3.4.4.1. FATHMM¶
Functional Analysis through Hidden Markov Models (FATHMM)[123] is a tool for predicting the functional effects of protein missense mutations using sequence conservation information (via HMMs), which can be (optionally) weighted by how likely a mutation in a protein/domain would be to lead to disease. FATHMM can only score missense mutations because it scores SNPs based on the probability of specific amino acids existing in proteins. Weightings are calculated from the frequency of disease-associated and functionally neutral amino acid substitutions in protein domains from human variation databases (the Human Gene Mutation Database[124] and Uniprot-KB/Swiss-prot[78]).
Consequence files describing whether an amino acid results in a missense, nonsense or synonymous SNP must first be obtained by using Ensembl’s Variant Effect Predictor[125] in order to create input to FATHMM. FATHMM then calculates conservation scores which are a measure of the difference in amino acid probabilities for a SNP according to the HMM, i.e. between a wild type amino acid and it’s substitution. A reduction in amino acid probabilities is interpreted as a prediction of deleteriousness (likelihood to cause harm), and the larger the reduction the greater the predicted harm.
3.4.5. Prediction tasks: Phenotype prediction¶
Phenotype prediction is the task of predicting phenotypes from genotypes. Specific tests of phenotype prediction might look like matching genotypes to profiles of traits, or predicting specific phenotypes from genotypes.
Although it’s often presented as a separate task, phenotype prediction is closely linked to protein function prediction. When variants on the protein-coding genome are known to be responsible for phenotypes, the assumption is that variant impacts the protein and the protein has a function that causes the phenotype when it behaves differently than usual. This is the assumption that underlies annotations between genes or proteins and phenotype terms, and it’s also the assumption that underlies phenotype “prediction” algorithms like 23andMe or Promethease’s health reports, which count the presence or absence of individual variants thought to be associated with disease, in order to inform potential phenotypes e.g. “You have 2 alleles associated with causing Breast Cancer”.
3.4.5.1. CAGI¶
Critical Assessment of Genome Intepretation[126] (CAGI) is a prediction competition open to the research community, in the same tradition as CAFA, this time aiming to objectively assess predictive methods for determining the phenotypic impacts of genomic variation across a number of different challenges.
The precision of the best methods in phenotype prediction of rare illnesses is still below 50%[127].